

Abstract
Background: FLT3-ITD mutation remains a great challenge in treating acute myeloid leukemia (AML) patients, for the high incidence of early relapse and drug-resistant mutations to tyrosine kinase inhibitors (TKIs). The FLT3-ITD mutation results in a significant and sustained activation of downstream signaling pathways. However, the physiological negative regulators of those aberrantly activated pathways have not been fully elucidated. In this study, we aim to identify and exploit a key molecule directly interacting with FLT3 receptor and down-regulating FLT3 associated pathways.
Methods: The potential proteins interacting with FLT3 were immunoprecipitated (IP) and screened by mass spectrometry. The binding of phosphatase SHP-1 and FLT3 was verified using co-IP and in situ proximity ligation assay (PLA) in cell lines and primary samples. The phosphatase activity and phosphorylation level of SHP-1 was determined by pNPP phosphatase activity assay and western blot. Biological effects of overexpressed SHP-1 by lentiviral vectors in AML cell-lines were assessed using proliferation, apoptosis and clonal formation assays in vitro and xenografts models in vivo. Plasmids of truncated or mutated structure of SHP-1 were constructed and overexpressed in cells to identify the key domain responsible for binding with FLT3 receptor. An artificial trans-membrane peptide was designed to activate SHP-1 activity and promote apoptosis in AML cells.
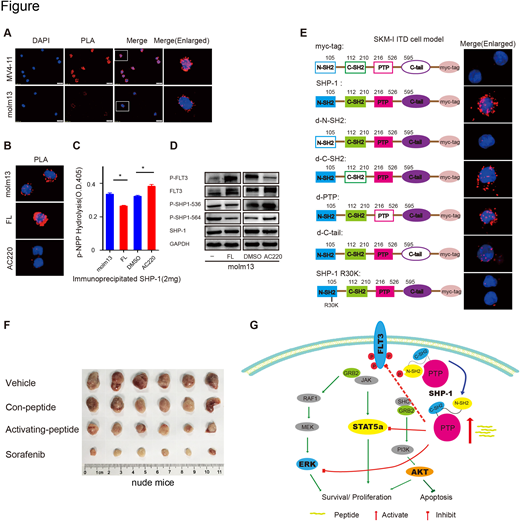
Results: The researchers performed immunoprecipitations (IP) followed by LC-MS/MS based screen in human leukemia cell line MV4-11 for three times, and found that the phosphatase SHP-1 binds to FLT3. In addition, the interaction between SHP-1 and FLT3-WT or FLT3-ITD was verified by co-IP and in situ proximity ligation assay (PLA) in AML cell lines, primary samples and overexpression model of 293-FT.Partial results are showed in Figure A. Furthermore, in heterozygous cell line molm13 and engineered FLT3-ITD knock-in cell line SKM-1 by crispr/cas9 techonology, combined with PLA, pNPP phosphatase activity assay and western blot, we found that SHP-1 may bind with p-FLT3 and acquired phosphatase activity(Figure B-D). In turn, p-FLT3 itself and some important proteins in the downstream pathway would be dephosphorylated and the signal transmission would be terminated. In FLT3-ITD cells, the phosphorylation level of SHP-1 significantly reduced.
By using lentivirus to overexpress or knockdown shp-1 protein in FLT3-WT and FLT3-ITD cells, the biological effects were studied. Experiments on cell apoptosis, proliferation and clonal formation have shown that overexpression of SHP-1 in AML cells may promote apoptosis, inhibit proliferation and clone formation, especially in FLT3-ITD cells. On the contrary, inhibition of shp-1 could promote proliferation and increase the ability of clone formation. Furthermore, DOX-inducible overexpression cell line of SHP-1 was obtained by tet-on virus system and the western blots showed that SHP-1 may inhibit p-ERK and p-STAT5 activity. In vivo experiments of NCG mice showed that, the degree of leukemia infiltration in peripheral blood was decreased in AML transplantation mice induced by DOX to express SHP-1 when compared to control group.
Next, according to the analysis of protein structure by computer simulation, we constructed truncated and mutated plasmids and overexpressed in cells, then using PLA to verify the binding , we found that FLT3 interact with N-SH2 domain of SHP-1,exactly the R30 site(Figure E). Therefore, by using specific trans-membrane peptide, the researchers found that it could activate SHP-1 and promote apoptosis in AML cells, especially FLT3-ITD ones, inhibit cell proliferation and the downstream signal pathway of FLT3.Lastly,injection of peptide in subcutaneous tumor model of nude mouse showed that the tumor was significantly smaller than that of the control group(Figure F).
Conclusion: We identify a negative regulator, namely phosphatase SHP-1, which directly interacts with FLT3 receptor through the N-SH2 domain and down-regulates FLT3 downstream signaling pathways. Overexpression of SHP-1 promotes apoptosis and inhibits proliferation in cells with FLT3-ITD mutation. Artificial trans-membrane peptides activating SHP-1 can reverse FLT3 associated aberrant signaling, thus providing a new potential strategy to target FLT3-ITD in AML. The possible mechanism is showed in figure G.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal